La introducción de los azoles cambió el tratamiento de la enfermedad fúngica. El ketoconazol se introdujo en 1985, pero en las dos siguientes décadas aparecieron el fluconazol, el itraconazol y el voriconazol. La información sobre su disponibilidad y coste en diferentes países se puede consultar en: “antifungalsavailability.org (GAFFI)”.
La baja toxicidad de estos agentes en comparación con la anfotericina B y su formulación oral transformó el tratamiento. También existen azoles tópicos utilizados, principalmente en el tratamiento de la candidosis superficiales, pero no todos están incluidos aquí.
Los azoles interfieren con la biosíntesis del ergosterol al inhibir la 14-α desmetilasa. Para obtener más información sobre este proceso y el desarrollo de resistencia, consulte Doctor Fungus o las reseñas de Cowen et al (2015) o Shafiei et al (2020).
Se recomienda la monitorización terapéutica del fármaco (TDM por sus siglas en inglés) cuando se prescribe voriconazol o itraconazol a determinados grupos de pacientes. Obtenga más información viendo nuestro seminario web de TDM y preguntas y respuestas en YouTube.
Los azoles interactúan con muchos otros medicamentos y pueden requerir ajustes de dosis. Obtenga más información utilizando la base de datos y la aplicación Interacciones de antimicóticos producidas por Fungal Infection Trust.
Los azoles orales tienen efectos dañinos en el feto al interferir con la esteroidogénesis. Sin embargo, en algunos casos faltan datos de ensayos en humanos (categoría C) y en otros casos, su uso aún puede estar indicado para infecciones fúngicas graves/invasoras (categoría D). Las formulaciones lipídicas de anfotericina B son generalmente una opción más segura durante el embarazo.
Obtenga más información en nuestra conferencia clínica sobre el uso de antimicóticos durante el embarazo y la lactancia.
- En ausencia de una alternativa adecuada se podría administrar fluconazol después del primer trimestre del embarazo
- Voriconazol está contraindicado durante el embarazo
- La anticoncepción eficaz es necesaria durante y hasta dos meses después de la interrupción del tratamiento con itraconazol

Conferencias y videos
Click here to download the slide decks for the lectures
Azole resistance
Azole resistance is becoming more common (perhaps due to heavy use of azoles in agriculture) so it is important to carry out susceptibility testing when possible, either by the CLSI microdilution method or using molecular methods to detect mutations in genes such as cyp51A.
Resistance may also arise in patients on long-term therapy for infections such as CPA. Antifungal stewardship programmes can help to guide usage to minimise this.
- Direct detection of some resistance mutations in Aspergillus fumigatus is possible with PCR (White et al, 2017; Dannaoui et al, 2017)
- Read about how researchers at MRCM used pyrosequencing to detect azole resistance in Aspergillus fumigatus strains isolated from CPA/ABPA patients at the National Aspergillosis Centre (van der Torre et al, 2020)
Folletos informativo
Posaconazol
| El nombre Posaconazol Noxafil (Merck) El posaconazol (Noxafil, MSD) es un triazol sintético soluble en grasas con una estructura química similar al itraconazol. Los estudios clínicos comenzaron en 1998 y fue autorizado su uso en humanos en 2006, para la profilaxis de las infecciones fúngicas en la leucemia aguda y en los transplantados de progenitores hematopoyéticos y como tratamiento de segunda línea en la aspergilosis invasora y la mucormicosis. Tiene el mismo mecanismo de acción de todos los azoles, que consiste en la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membrana celular de los hongos. |
| Dosis y vías de administración La solución oral de posaconazol contiene numerosos ingredientes como polisorbato 80 y sabor a cerezas. Asimismo, hay tabletas y solución intravenosa. La solución oral se absorbe mejor cuando se administra con comida, ya que la misma aumenta su biodisponibilidad intestinal. Las tabletas se pueden tomar a cualquier hora. La dosis estándar de la solución oral es de 300 mg/12h, el primer día para tomar una sola dosis de 300 mg/día subsiguientemente. En pacientes que están recibiendo el posaconazol en tabletas como profilaxis, la dosis es de 300 mg/12h el primer día para seguir con 300 mg/día los subsiguientes. La concentración de posaconazol en la suspensión oral es de 40 mg/ml, así que una cuchara de 5 ml supone una dosis de 200 mg. Para las tabletas y la solución intravenosa, la dosis estándar es de 300 mg/12h el primer día para seguir con 300 mg/día administrada en una sola dosis. |
| Hongos sensibles al posaconazol Tiene una gran espectro de acción y así es activo frentea Aspergillus spp., Candida spp., Coccidioides spp., Histoplasma, Paracoccidioides brasiliensis, Blastomyces dermatitidis, Cryptococcus spp., Sporothrix schenckii, varias especies de Mucorales, Fusarium spp., hongos negros como Bipolaris spp. and Exserohilum spp. Posaconazol es fungicida en el laboratorio contra la mayoría de las cepas de Aspergillus a concentraciones clínicamente relevantes. Se ha descrito resistencia adquirida al posaconazol en Aspergillus fumigatus y Candida albicans pero no es frecuente. |
| Dosificaciones habituales Para el tratamiento de las infecciones fúngicas refractarias al tratamiento, aspergilosis, zigomicosis u otras infecciones raras, se administran 300 mg/12 horas el primer día para seguir con 300 mg/día los subsiguientes. La monitorización de las concentraciones sanguíneas de posaconazol ha demostrado que varían 10 veces y que las concentraciones bajas se asocian con fallos terapéuticos (Un aumento de la dosis a 400 mg/8 horas puede ser beneficioso, si la razón de las bajas concentraciones no es una interacción farmacológica). Para la profilaxis de la infección fúngica, es preferible una dosis de 300 mg IV/12 horas el primer día, seguidos de 300 mg/día los subsiguientes. Las tabletas por vía oral se administran a dosis de 300 mg/12 horas el primer día, seguidos de 300 mg/día los subsiguientes. Para la suspensión oral la dosis recomendada es de 200 mg/8 horas. Para la candidosis oral, 100 mg/12h de dosis de carga seguidos de 100 mg/día. Cuando la infección ha sido refractaría al itraconazol o el fluconazol se recomienda que la dosis de la suspensión oral de posaconazol sea de 400 mg/12 horas (consultar las recomendaciones del EMC o Medscape) |
| Metabolismo, distribución y excreción El posaconazol tarda en absorberse entre 3 y 5 horas. Un aumento de dosis desde los 200 hasta los 800 mg incrementa la exposición de forma proporcional. Se tarda entre 7-10 días en alcanzar la fase de equilibrio en las concentraciones sanguíneas, pero la mayoría de los pacientes tiene concentraciones terapéuticas en los dos primeros días. El posaconazol, aparentemente tiene un gran volumen de distribución, lo que sugiere una amplia repartición tisular. También tiene una unión a proteínas muy elevada (>98%). Se metaboliza primariamente por el hígado en conjugados glucurónidos. La vida media de eliminación es de 35 horas con la mayoría del fármaco eliminado por heces sin metabolizar. La variación interindividual en la absorción y las consiguientes concentraciones es grande. Algunos pacientes transplantados tienen bajas concentraciones sanguíneas de forma consistente. |
| Interacciones Tiene menos interacciones que el itraconazol o el voriconazol. Las concentraciones sanguíneas disminuyen cuando se administran conjuntamente con inductores de enzimas como la rifampicina, rifabutina, efavirenz, fenitoina, fenobarbitona, carbamazepina, etc. Se absorbe menos cuando se administra conjuntamente con inhibidores de la bomba de protones como omeprazol, lansoprazol, etc., o antagonistas de los receptores H2 como la cimetidina, ranitidina, etc. Consultar la base de datos de interacciones. El posaconazol es un potente inhibidor del CYP3A4. Esto significa que probablemente se elevarán las concentraciones sanguíneas de los fármacos que son metabolizados por este sistema. Ejemplos importantes son la ciclosporina, tacrolimus, sirolimus, terfenadina, astemizol, cisaprida, ergotamina (anti migrañoso) , simvastatina, lovastatina, atorvastatina (estatinas), vincristina, vinblastina (qimioterápicos), midazolam, diazepam (benzodiacepinas) y algunos inhibidores de la proteasa del HIV. Otros fármacos con menores interacciones con el posaconazol son la digoxina y la glipizida para la diabetes, y ciertos bloqueadores de los canales del calcio como la nifedipina y el diltiazem. |
| Efectos adversos Laos más frecuentes incluyen nauseas, diarrea moderada, malestar abdominal, indigestión y boca seca. Los tests de función hepática pueden estar alterados con la misma frecuencia que cuando se administra fluconazol. Las erupciones cutáneas por posaconazol son relativamente frecuentes. Fiebre, fatiga, falta de energía y pérdida de apetito también suelen aparecer. Otro efectos secundarios como disminución de los leucocitos o plaquetas se asociaron con el posaconazol porque se estudió en pacientes con neutropenia y en tratamiento contra la leucemia. La frecuencia de estos efectos en pacientes sin leucemia es baja. Otros efectos menores o incluso significativos pueden ocurrir. Ver el video “Reacciones cutáneas de los antifúngicos”. |

Voriconazol
| El nombre Voriconazol V-fend (Pfizer); voriconazole Accord (Accord Healthcare). El voriconazol (Vfend, Pfizer) fue autorizado en el año 2002, para el tratamiento de la aspergilosis invasora, de las infecciones por Fusarium y Scedosporium y de la candidosis resistente. Se define como un triazol de segunda generación. Estructuralmente se parece al fluconazol, pero su farmacocinética y espectro antifúngico es completamente diferente. Tiene el mismo mecanismo de acción de todos los azoles, que consiste en la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membrana celular de los hongos. |
| Dosis y vías de administración De voriconazol, están disponibles preparaciones oral, intravenosa y ocular. Por vía intravenosa en adultos y para infecciones potencialmente mortales se aconseja una dosis de carga de 6 mg/kg/12 horas seguidos de 4 mg/kg/12 horas. Las dosis aconsejadas en niños son de 7 mg/kg/12 horas, aunque los niños pequeños pueden requerir dosis más elevadas, debido a su rápida eliminación. Si se pueden monitorizar las concentraciones sanguíneas, la dosis pueden ser ajustadas en base a los resultados de la prueba. La biodisponibilidad es cercana al 100% por lo que se puede confiar en la vía oral. La dosis recomendada es de 200 mg/12 horas. Se deben dar dosis menores a adultos que pesen menos de 40 kg, a los ancianos y a cualquier paciente con enfermedad hepática para evitar concentraciones que puedan causar toxicidad. Debido a que la farmacocinética del voriconazol en adultos no es lineal, el aumento de las dosis puede causar una desproporcionada exposición al voriconazol. En niños esto no ocurre. |
| Hongos sensibles al voriconazol Tiene un amplio espectro de actividad. Es activo frente a la inmensa mayoría de especies de Candida spp., Cryptococcus spp., todas las especies de Aspergillus, Scedosporium apiospermum, algunas cepas de Fusarium y otros patógenos raros. No es activo frente a los Mucorales como Mucor spp., Rhizopus spp., Rhizomucor spp., etc. |
| Dosificaciones habituales Aspergilosis invasora: en adultos, la dosis de carga es de 6 mg/kg/12 horas, seguida de 3-4 mg/kg/12 horas por vía intravenosa y después 200 mg/12 horas por vía oral. En niños las dosis son proporcionalmente mas grandes. |
| Metabolismo, distribución y excreción El voriconazol se metaboliza en el hígado. Las enzimas más importantes que participan en el metabolismo son CYP 2C19, CYP 3A4 and CYP 2C9. La variación genética interindividual, particularmente con CYP 2C19, afecta parcialmente la velocidad con la que el voriconazol se metaboliza. Aproximadamente el 3% de los caucasianos, el 20% de los japonés y otros asiáticos metabolizan lentamente el voriconazol. Estos pacientes pueden acumular el voriconazol y, en ocasiones, se alcanzan concentraciones muy elevadas en sangre. Los ancianos y los que tienen enfermedad hepática moderada o grave también metabolizan lentamente el voriconazol. Los niños pequeños metabolizan el voriconazol muy rápido y los niños mayores y adolescentes, rápido por lo que es necesario aumentar las dosis en estas edades. |
| Interacciones Como con otros azoles hay un numero sustancia de interacciones. La rifampicina, rifabutina (en menor cantidad), fenitoina y carbamazepina reducen las concentraciones de voriconazol. El voriconazol aumenta las concentraciones de algunos antihistamínicos (terfenadina y astemizol) prednisolona, sirolimus, ciclosporina (mitad de la dosis), tacrolimus (reducir la dosis un tercio), ergotamina, warfarina, sulfonilureas, estatinas, midazolam y otras benzodiacepinas, vincristina y algunos fármacos anti HIV. Unos pocos fármacos reducen el metabolismo del voriconazol incluyendo omeprazol y probablemente otros inhibidores de las bombas de protones. Ver la base de datos de interacciones. |
| Efectos adversos El efecto secundario más frecuente del voriconazol, quizás sean los efectos visuales. En los primeros días tras la administración del voriconazol, los pacientes describen líneas onduladas, visión borrosa moderada o luces brillantes que típicamente duran unos pocos minutos. Una investigación cuidadosa no ha sido capaz de descubrir cual es el mecanismo de este efecto. Suelen desaparecer con el tiempo y rara vez han producido un daño permanente en los ojos. En algunos pacientes, las pruebas de función hepática están alteradas y cuando están llamativamente elevadas hay que interrumpir el voriconazol. Cuando las concentraciones sanguíneas son elevadas, el paciente se queja de embotamiento y falta de concentración. En ocasiones hay alucinaciones y franca confusión. En una minoría de pacientes y tras meses de tratamiento puede aparecer neuropatía periférica. En pacientes con concentraciones muy elevadas de voriconazol han aparecido efectos secundarios graves. El tratamiento prolongado con voriconazol puede dar lugar a un enrojecimiento de la piel expuesta al sol, especialmente la cara, con sequedad de ojos y labios, especialmente a los individuos de piel blanca. Se aconseja crema protectora solar SPF 50, así como evitar ambientes soleados. En los coches se puede utilizar filtros solares en las ventanas. Ver el video “reacciones cutáneas a los antifúngicos” y “Toxicidad neurológica“. |
| Otra información Una información detallada está disponible en la sección de tratamiento de la página WEB de Aspergillus. |

Itraconazol
| El nombre Itraconazol Sporanox (Janssen); TOLSURA/Lozanoc (Mayne Pharma) El itraconazol (Sporanox, Janssen) es un antifúngico triazol sintético soluble en grasa. Tiene el mismo mecanismo de acción de todos los azoles, que consiste en la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membrana celular de los hongos. Los estudios con el itraconazol comenzaron en 1984 y su uso clínico fue autorizado en 1991. El antifúngico ya no tiene patente, pero para las formulaciones de las soluciones oral e intravenosa la patente sigue en vigor. |
| Dosis y vías de administración Hasta 1997 solo estuvieron disponibles las cápsulas, que se absorben mejor si se toman con comida. Posteriormente se introdujo una solución líquida, especialmente para tratar la candidosis oral resistente al fluconazol en pacientes con SIDA. También está disponible una solución intravenosa en ciclodextrina. Hay variaciones entre la biodisponibilidad de diferentes formulaciones de cápsulas y se han detectado fallos terapéuticos al cambiar entre ellas. La solución oral se usa, típicamente para el tratamiento de la candidosis oral, especialmente en pacientes con SIDA y en la profilaxis de la infección fúngica en leucemias o tras el transplante de progenitores hematopoyéticos. También se utiliza cuando los pacientes están recibiendo medicación para disminuir la acidez en el estómago. La suspensión oral se absorbe mejor cuando el estómago está vacío. El itraconazol intravenosos se suele administrar a dosis de 5 mg/kg/12h y está indicado en pacientes que no pueden tomar medicación oral o con grave enfermedad. |
| Hongos sensibles al itraconazol El itraconazol es uno de los antifúngicos con mayor espectro de actividad, incluyendo Aspergillus spp, Blastomyces dermatitidis, Candida (todas las especies y la mayoría de las cepas incluyendo algunas con resistencia al fluconazol), Coccidioides spp., Cryptococcus spp., Histoplasma spp, Paracoccidioides brasiliensis, Scedosporium apiospermum and Sporothrix schenckii. También es activo frente a los dermatofitos. No es activo frente a los Mucorales, Fusarium spp. y otras hongos raros. Hasta el desarrollo del posaconazol, fue el antifúngico más activo frente a los hongos negros incluyendo Bipolaris, Exserohilum, etc. Se ha descrito resistencia al itraconazol en Candida spp., Aspergillus fumigatus y A. niger. |
| Dosificaciones habituales Para la mayoría de las vaginitis, dos dosis de 100 mg de itraconazol son suficientes. Entre 5 y 7 días de tratamiento son suficientes para la candidosis oral. Ocasionalmente, en pacientes con SIDA con cepas resistentes al fluconazol, es necesario un tratamiento continuado. Para la mayoría de las infecciones cutáneas, 7 días de tratamiento es suficiente, excepto para la tiña pedis extensa (infección por dermatofitos de los pies y los espacios interdigitales) y algunas infecciones del cuero cabelludo donde se suelen requerir 3 semanas de tratamiento. Para la onicomicosis de las manos, se recomienda 3 meses de tratamiento, y para la de los pies, 6 meses, a veces con administración discontinua (lo que se conoce en inglés como “pulse therapy”) Para infecciones graves y potencialmente mortales, como la aspergilosis invasora, se recomiendan dosis de carga de 200 mg, 3 o 4 veces al día durante 3 o 4 días. Para la aspergilosis invasora, se recomienda la utilización de 400 mg/día o incluso una dosis mayor durante, por lo menos, 3 meses pero a menudo se alcanza el año de tratamiento o incluso más. En la aspergilosis broncopulmonar alérgica (ABPA), una dosis de 400 mg/día parece ser ligeramente superior a una de 200 mg/día, pero la de 200 mg tienen un indudable efecto. El tratamiento se debe mantener durante un mínimo de 3 meses, ya que cuando se interrumpe la recaída es frecuente, y por lo tanto, un tratamiento más prolongado o incluso continuo puede ser apropiado. El itraconazol no es muy efectivo en la aspergilosis pulmonar crónica, pero a dosis de 400 mg/día durante muchos meses tiene un efecto parcial. Se recomienda la monitorización de las concentraciones sanguíneas del itraconazol para asegurar que el paciente tiene concentraciones terapéuticas, lo que además ayuda a comprobar el seguimiento del tratamiento y minimizar la toxicidad. Para la histoplasmosis, paracoccidioidomicosis, blastomicosis y esporotricosis, a dosis de 200 mg/día durante 3-6 meses es efectivo en la mayoría de los casos. Si el paciente tiene SIDA, se recomienda tratar estas infecciones con dosis mayores, del orden de 400 mg/día. El tratamiento puede ser de por vida, aunque se puede reducir a 200 mg/día, una vez que la infección se considere controlada. Para la coccidioidomicosis, una dosis de 400 mg/día parece ser la más apropiada. Para las infecciones de los senos paranasales por hongos negros se han utilizado dosis mas elevadas, entre 400-1200 mg/día, pero se aconseja consultar con un especialista. |
| Metabolismo, distribución y excreción La absorción intestinal del itraconazol es incompleta. El itraconazol sufre una gran metabolización y uno de ellos, el hidroxi itraconazol, tiene actividad antifúngica. Esta muy unido a proteínas y por tanto no alcanza el LCR o la orina. Sin embargo, se une a la queratina y por tanto, se encuentran en grandes concentraciones en la piel y las uñas. La vida media del itraconazol depende de la dosis, pero típicamente es entre 48 a 60 horas. Se tarda unas dos semanas en alcanzar concentraciones sanguíneas de equilibrio. Si se utilizan dosis de carga, la situación de equilibrio puede alcanzarse en una semana. |
| Interacciones El itraconazol tienen mucha interacciones lo que es una de sus limitaciones. Cuando se dan antiácidos se reduce la absorción intestinal de las cápsulas, pero aumenta cuando es la solución lo que se administra. Las concentraciones sanguíneas se reducen significativamente cuando se administran inductores de enzimas como la rifampicina, la fenitoina o la carbamazepina, y por tanto, el tratamiento puede fracasar. Asimismo, el itraconazol impide la metabolización de ciertos fármacos como la terfenadina, el astemizol y el cisaprida lo que puede causar arritmias cardíacas graves, y por tanto, se debe evitar administrar estos fármacos con itraconazol. Aumenta los efectos de algunos sedantes, especialmente del midazolam y triazolam. El itraconazol también puede aumentar las concentraciones sanguíneas de la digoxina, ciclosporina y warfarina por lo que estos fármacos y sus efectos deben ser cuidadosamente monitorizados. La administración conjunta del itraconazol con la vincristina prolonga la acción de esta última y puede causar daño neurológico, que es generalmente temporal, pero puede ser muy problemático. Las interacciones con esteroides inhalados son importantes, y pueden producir fallo adrenal permanente. Consultar la base de datos de interacciones medicamentosas. |
| Efectos adversos El itraconazol se suele tolerar adecuadamente. Los efectos secundarios más frecuentes son nauseas, mareo, malestar abdominal y estreñimiento. Estos efectos secundarios, pero diarrea son más frecuentes con la solución. En ancianos la retención de líquidos puede ser un problema. Erupciones cutáneas ocurren, aproximadamente, en 1 de 20 pacientes y en ocasiones, pueden ser graves. Ocasionalmente se producen alteraciones hepáticas, que en casos raros, pueden llegar a ser graves con ictericia. También puede aparecer, en ocasiones, supresión de la glándula adrenal, concentración baja de potasio e hipertensión. En algunos pacientes puede aparecer una neuropatía periférica y temblores. Ver el video “Reacciones cutáneas a los antifúngicos” y el video “Toxicidad neurológica” |

Fluconazol
| El nombre Fluconazol Diflucan (Pfizer) El fluconazol (Diflucan, Pfizer) es un compuesto sintético conocido como un bis triazol. Se utilizó, por primera vez, en pacientes en 1994/95 y su uso fue autorizado en 1990. Su patente expiró en 2003. Su mecanismo de acción es la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membrana celular de los hongos. |
| Dosis y vías de administración Está disponible en cápsulas, suspensión oral e infusión intravenosa. La dosis recomendada para la candidosis oral es 100 mg/día. Para la vaginitis, la dosis es de 150 mg/día. Para la prevención de la infección fúngica en leucémicos se han utilizado y analizado dosis entre 50-400 mg/día. La mayoría de los expertos recomiendan entre 100 y 400 mg/día. Para el tratamiento de la candidemia y otras infecciones graves por Candida, se recomienda una dosis mínima de 400 mg/día, tras una dosis de carga de 800 mg. En pacientes en hemofiltración y/o recibiendo rifampicina, se recomienda aumentar las dosis (ver más abajo). Algunos autores han sugerido que una dosis de 800 mg/día es superior para la candidemia, pero los datos no son convincentes. Para el tratamiento de la meningitis criptocócica, tras el tratamiento inicial con anfotericina B, se utiliza una dosis mínima de 400 mg/día hasta que el paciente está estable para luego pasar a 200 mg/día. Dosis mas elevadas, entre 800-1200 mg/día, se utilizan como tratamiento primario alternativo (es preferible combinarlo con fluorocitosina) o en el caso de fracaso del tratamiento primario. Para el tratamiento de la meningitis por Coccidioides, se han utilizado dosis elevadas entre 800-1200 mg/día. El resto de tratamientos, prácticamente, utiliza dosis entre 100-400 mg/día. Dosis más reducidas pueden ser apropiadas cuando existe una disfunción renal grave. |
| Hongos sensibles al fluconazol El fluconazol es activo frente a la mayoría de las especies de Candida, exceptuando a Candida krusei que es intrínsecamente resistente y Candida glabrata que es capaz de desarrollar resistencia secundaria con facilidad. Algunas cepas de Candida albicans, Candida tropicalis, Candida parapsilosis han desarrollado resistencia secundaria pero la tasa no es alarmante. Para otras especies, es importante verificar la sensibilidad al fluconazol mediante un método estandarizado. Es también activo frente la gran mayoría de las cepas de Cryptococcus spp. También tiene actividad frente a Trichosporon spp., Rhodotorula rubrum y los hongos dimórficos endémicos como Blastomyces dermatitidis, Coccidioides spp., e Histoplasma spp. sin embargo, es menos activo que el itraconazol frente a estos últimos hongos dimórficos. No tiene actividad contra Aspergillus spp. o Mucorales. Tiene cierta actividad frente a los dermatofitos como Trichophyton spp. Se ha documentado resistencia al fluconazol en Candida albicans, especialmente en aislados orales de pacientes con SIDA tratados con fluconazol. En un hospital general, las tasas típicas de resistencia de Candida albicans al fluconazol están entre el 3-6%. |
| Metabolismo, distribución y excreción El fluconazol es soluble en agua, se absorbe bien tras la administración oral y las concentraciones séricas son equivalentes a la dosis administrada. Los bebes y los niños, especialmente con fiebre, tienen un metabolismo acelerado. La mayoría del fluconazol se excreta por la orina. Una fracción muy pequeña se metaboliza en el hígado. Penetra bien en el cerebro, los ojos, la saliva y la orina. Un máximo del 17% de la dosis se excreta en la leche materna en mujeres lactantes, con efectos secundarios mínimos o nulos en los bebés expuestos a ella. |
| Interacciones Existen pocas interacciones del fluconazol con otros fármacos. La mas llamativa es con la rifampicina y la fenitoina. Ambas aceleran el metabolismo del fluconazol en el hígado, con disminución de sus concentraciones sanguíneas. Además el fluconazol puede elevar las concentraciones séricas de la fenitoina y los antidiabéticos como clorpropamida, glibenclamida, glipizida y tolbutamida así como la warfarina. También aumenta levemente las concentraciones de la ciclosporina. Ver la base de datos de interacciones |
| Efectos adversos El fluconazol es, en general, bien tolerado. Probablemente es el antifúngico menos tóxico de los que se usan sistémicamente. Los efectos secundarios mas frecuentes son nauseas y malestar abdominal, y en ocasiones hay elevación de las enzimas hepáticas. Una erupción cutánea, que en ocasiones puede ser grave, aparece en 1 de cada 20 pacientes tratados. El fluconazol no tiene efectos endocrinos. No debe ser utilizado en pacientes embarazadas. |

Ketoconazol
| NAMES Ketoconazol El ketoconazol es el primer azol oral. Es un imidazol sintético. Se estudió por primera vez a principios de los 80 y su uso clínico se autorizó en 1985. Como todos los azoles, su mecanismo de acción es la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membrana celular de los hongos. |
| Dosis y vías de administración Está disponible en tabletas para administrar por vía oral y en preparaciones tópicas como el champú Nizoral. |
| Hongos sensibles al ketoconazol Es activo frente Candida spp, Blastomyces dermatitidis, Coccidioides spp, Histoplasma and Paracoccidioides. Es también activo frente a los dermatofitos, pero no tanto como el itraconazol o la terbinafina. No tienen actividad frente a Aspergillus spp. |
| Dosificaciones habituales El ketoconazol se utiliza en el tratamiento de la candidosis oral, a dosis de 200 mg/día o a 400 mg/día si el paciente tiene SIDA y hay cepas resistentes. Para la blastomicosis, histoplasmosis, coccidioidomicosis y paracoccidioidomicosis, se han utilizado dosis de 200 mg/día/12horas, pero con resultados inferiores que con el itraconazol. La misma dosis se usa para las infecciones cutáneas. Como el ketoconazol no es tan activo como el itraconazol, los cursos de tratamiento para las micosis endémicas son más prolongados. |
| Metabolismo, distribución y excreción Se absorbe bien tras la administración oral y la comida no tiene ningún impacto en la misma. Se metaboliza, casi completamente, en el hígado y los metabolitos se eliminan por la bilis. |
| Interacciones Las concentraciones sanguíneas de ketoconazol son ligeramente más bajas si el paciente está recibiendo antiácidos. La rifampicina aumenta el metabolismo del ketoconazol y por tanto disminuye su concentración. El ketoconazol no se debe administrar conjuntamente con terfenadina, astemizol y cisaprida ya que aumenta sus concentraciones, lo que puede inducir la aparición de arritmias cardíacas (QT prolongado). También puede prolongar la acción de algunos sedantes, como el midazolam y el triazolam. Aumenta las concentraciones sanguíneas de la ciclosporina, y de hecho se ha utilizado en algunos centros de trasplantes para disminuir el coste de la ciclosporina. También prolonga el efecto de la warfarina. Consultar la base de datos de interacciones. |
| Efectos adversos Los efectos secundarios más frecuentes son pérdida de apetito, sensación de malestar general y vómitos y ocurren en 1 de cada 10 pacientes. Son particularmente problemáticos a dosis mayores de 400 mg/día. En 1 de cada 20 pacientes produce ligeras elevaciones de las enzimas hepáticas pero en ocasiones la afectación del hígado ha sido grave. Si el tratamiento se prolonga más de dos semanas, hay un efecto adverso raro (1-10.000-15.000) que es fatal si no se diagnostica y se interrumpe el ketoconazol. El ketoconazol también inhibe la síntesis de testosterona por la glándula adrenal y por los testículos, especialmente si las dosis son elevadas. Como consecuencias aparecen pérdida de peso, aumento de pecho y ausencia de deseo sexual. Existe la posibilidad de que esto efectos sean más problemáticos en los adolescentes y por eso, si es posible, se debe de evitar su administración en este grupo de edad. Tampoco se debe utilizar en el embarazo. El ketoconazol, a dosis de 600 mg/día, es bastante efectivo y se puede utilizar para controlar el exceso de corticoesteroides asociado al síndrome de Cushing. Recientemente la “Food and Drug Administration” (FDA) ha restringido la utilización del ketoconazol como un fármaco de último recurso, ya que existen otros antifúngicos que son superiores y con menos efectos adversos (Julio, 2013). Vínculo con la resolución. |

Econazol
| El nombre Econazol El econazol (Ecostatin, Ecoderm, Pevaryl y otros muchos nombres) es un imidazol sintético con una estructura similar al miconazol. Fue sintetizado por primera vez en 1969, usado en pacientes en los 70 y autorizado para uso clínico en 1974. Su mecanismo de acción es la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membrana celular de los hongos. Estudios realizados con Candida albicans sugieren que puede tener también otro tipo de acciones celulares. |
| Dosis y vías de administración Solo se usa tópicamente como 1% crema, loción, solución o polvos, 3 veces al día durante 2-4 semanas. El econazol se utiliza para la dermatitis seborreica, el eczema del pañal debido a Candida, algunos casos de tiña, como la cruris o el pie de atleta. Una solución al 1% se puede utilizar para la otitis externa. Para aquellas condiciones que cursan con inflamación, como la dermatitis seborreica, la combinación un corticosteroide tópico puede ser beneficiosa. Para la vaginitis por Candida, se pueden utilizar óvulos de 150 mg/día durante 3 días. Alternativamente, se puede aplicar cada noche 5g de una crema vaginal al 1% durante 2 semanas. La crema también se puede utilizar para tratar la balanitis. |
| Hongos sensibles al econazol Es activo frente la mayoría de las especies de Candida. También es activo frente a los patógenos cutáneos Trichophyton spp., Microsporum spp., Epidermophyton floccosum y Malassezia spp., así como frente a algunos patógenos sistémicos como Aspergillus spp. También tiene alguna actividad antibacteriana. Se han documentado algunas cepas resistentes de Candida albicans. Esta resistencia se puede inferir de los resultados de la sensibilidad frente a miconazol. |
| Metabolismo, distribución y excreción La absorción sistémica del econazol es mínima y aplicado en la piel penetra solo en la epidermis. Tras la aplicación vaginal se absorbe una mínima cantidad, pero que puede ser suficiente para desencadenar un ataque de porfiria aguda intermitente, en aquellos pacientes que tenga esta rara enfermedad. |
| Interacciones Hay pocas interacciones con el econazol. Los óvulos pueden dañar los preservativos de látex o los diafragmas y por tanto se aconseja tomar otras medidas contraceptivas adicionales. Ver la base de datos de interacciones. |
| Efectos adversos Puede producir irritación cutánea y sensación de quemazón. Se puede utilizar en el embarazo aunque se prefiere el uso de la nistatina. |

Clotrimazol
| El nombre Clotrimazol El clotrimazol (Canesten, Bayer; y otros muchos nombres) es un imidazol sintético. Se utilizó en pacientes por primera vez a finales de los 70 y su uso fue autorizado antes de 1982. En EEUU se autorizado su uso sin receta en 1989. Sus patentes, algunas con diferentes formulaciones, comenzaron a expirar a principios de los años 90. Su mecanismo de acción es la inhibición de la 14-alfa demetilasa, una enzima que participa en la síntesis de ergosterol, un componente principal de la membranas celular de los hongos. |
| Dosis y vías de administración Solo se utiliza de forma tópica. Se puede encontrar con otros compuestos como los corticosteroides. Las preparaciones más habituales son como crema, loción o spray al 1%, la cual se aplica 2 o 3 veces al día durante 2-4 semanas. El clotrimazol se utiliza para la dermatitis seborreica, el eczema del pañal debido a Candida, algunos casos de tiña como la cruris o el pie de atleta. Una solución al 1% se puede utilizar para la otitis externa. Para aquellas condiciones que cursan con inflamación, como la dermatitis seborreica, la combinación un corticosteroide tópico puede ser beneficiosa. Para la vaginitis por Candida, se pueden utilizar óvulos de 100 mg durante 6 días, de 200 mg durante 3 o de 500 mg como dosis única. Alternativamente, las cremas vaginales de 1%, 2% o 10% tiene las mismas dosis que los óvulos y pueden utilizarse con la misma duración. También se puede utilizar la crema para tratar la balanitis. Para tratar la candidosis oral se pueden utilizar pastillas de 10 mg. Se dejan disolver en la boca 5 veces al día durante 14 días. Para prevenir la candidosis oral se pueden dar 3 veces al día. |
| Hongos sensibles al clotrimazol Es activo frente la mayoría de las especies de Candida. También es activo frente a los patógenos cutáneos Trichophyton spp., Microsporum spp., Epidermophyton floccosum y Malassezia spp. In vitro también tienen actividad contra muchos hongos sensibles a los azoles como Coccidioides immitis pero como no se puede administra sistémicamente, esta actividad in vitro es poco importante. Se ha documentado resistencia de Candida albicans al clotrimazol, especialmente en cepas aisladas de candidosis oral en pacientes con SIDA tratados con este antifúngico. Las tasas típicas de resistencia en un hospital general don entre el 3 y el 6%. |
| Metabolismo y distribución Es prácticamente insoluble en agua. Aplicado en la piel penetra en la epidermis sin absorción sistémica. Tras la aplicación vaginal se absorbe sistémicamente entre el 3-10%. El clotrimazol es metabolizado en el hígado. |
| Interacciones Hay pocas interacciones con el clotrimazol. Los óvulos pueden dañar los preservativos de látex o los diafragmas y por tanto se aconseja tomar otras medidas contraceptivas adicionales. Ver la base de datos de interacciones. |
| Efectos adversos Es generalmente bien tolerado. Con las pastillas se han reportado, nauseas, vómitos, mal sabor de boca y picor. Con las cremas puede haber irritación cutánea y sensación de quemazón. Se puede utilizar en el embarazo aunque se prefiere el uso de la nistatina. |

Isavuconazol
| El nombre Isavuconazol El profármaco del isavuconazol (CresembaÒ) es el sulfato de isavuconazonio, un antifúngico azólico, que impide la síntesis de ergosterol, un compuesto necesario para la integridad de la membrana celular de los hongos. Su mecanismo de acción es a través de la inhibición de la 14-alfa-lanosterol demetilasa, una enzima dependiente del citocromo P-450. La FDA y la EMA han aprobado el isavuconazol para el tratamiento de la aspergilosis invasora y la mucormicosis invasora en pacientes de 18 años de edad o mayores. |
| Dosis y vías de administración El sulfato de isavuconazonio (profármaco del isavuconazol) está disponible para administración intravenosa y oral. Un vial intravenoso contiene 372 mg de sulfato de isavuconazonio equivalente a 200 mg de isavuconazol; Una cápsula oral contiene 186 mg de el sulfato de isavuconazonio equivalente a 100 mg de isavuconazol. La siguiente tabla explica detalla la dosificación del isavuconazol: 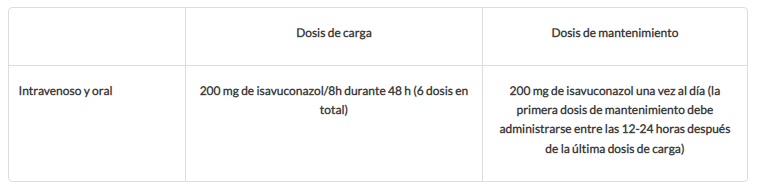 No es necesario dar una dosis de carga cuando se decide cambiar de la administración intravenosa a la oral. Se ha demostrado que ambas formulaciones son bioequivalentes. La edad, el sexo, la raza, la insuficiencia renal o la insuficiencia hepática leve o moderada no obligan a hacer ajustes en las dosis administradas. Sin embargo, no hay datos en pacientes pediátricos o en aquellos con insuficiencia hepática grave (Clase C de Child-Pugh). Tampoco hay datos en mujeres embarazadas y por tanto, no se recomienda su administración. Además se excreta en la leche de ratas lactantes, por lo que la lactancia materna está contraindicada mientras se administre el isavuconazol. Por vía intravenosa hay que administrarlo a través de un filtro con tamaño de poro entre 0,2-1,2 mm durante 1 hora. |
| Hongos sensibles al isavuconazol El isavuconazol tiene actividad in vitro contra Aspergillus fumigatus, A. flavus, A. terreus, A. niger y A. nidulans. Además, es activo contra Histoplasma capsulatum, Coccidioides immitis, Blastomyces dermatitidis, Candida spp, Cryptococcus spp y Trichosporon spp. Contra Mucorales. Fusarium spp and Scedosporium apiospermum su actividad es variable. También se ha demostrado su actividad contra los hongos que causan la feohifomicosis y la cromoblastomicosis. |
| Metabolismo, distribución y excreción En voluntarios sanos, y tras la administración oral de una o varias dosis del profármaco las concentraciones plasmáticas máximas se alcanzan entre 2 y 3 horas. La biodisponibilidad es del 98%. La comida no interfiere en la absorción del profármaco, el cual es absorbido y rápidamente hidrolizado a isavuconazol por esterasas. En el plasma no se detectan concentraciones significativas del profármaco o productos del metabolismo del mismo. El metabolismo primario es hepático. Es un substrato de las enzimas 3A4 y 3A5 del citocromo P450. Aparentemente tiene un gran volumen de distribución por lo que se distribuye ampliamente en los tejidos. Su unión a proteínas es mayor del 99%. La semivida de eliminación es de aproximadamente 130 horas y se requiere una dosis de carga para alcanzar la concentración estacionaria. La excreción se realiza principalmente por heces. Menos del 1% de la dosis administrada se elimina por orina. |
| Interacciones El isavuconazol tiene menos interacciones que otros azoles. Es un inhibidor del CYP3A4 por lo que si se administra con potentes inhibidores, como lopinavir/ritonavir, se pueden elevar significativamente las concentraciones plasmáticas del isavuconazol. Por el contrario, la coadministración con inductores del CYP3A4, como la rifampicina, carbamazepina, la hierba de San Juan, o los barbitúricos, pueden disminuir las concentraciones del isavuconazol. Asimismo, el isavuconazol disminuye las concentraciones de lopinavir/ritonavir y bupropión. El isavuconazol puede aumentar las concentraciones de atorvastatina, ciclosporina, sirolimus, tacrolimus, midazolam, micofenolato mofetil, y digoxina y por lo tanto puede ser necesaria la monitorización de sus concentraciones plasmáticas y/o el ajuste de las dosis. Consultar la base de datos de interacciones. |
| Efectos adversos Los efectos adversos mas frecuentes del isavuconazol en orden decreciente son nauseas, vómitos, diarrea, cefalea, pruebas hepáticas alteradas, hipokaliemia, estreñimiento, disnea, tos, edema periférico y dolor de espalda. Ver el video :Reacciones cutáneas de los antifúngicos |
Oteseconazol
| El nombre Oteseconazol (Vivjoa) |
| Aprobación Descubierto en 2013 como VT-1161 por Viamet Pharmaceuticals. Aprobado para uso en los Estados Unidos en 2022 para el tratamiento de la candidiasis vulvovaginal recurrente (CVVR), por Mycovia Pharmaceuticals. |
| Estructura y mecanismo Oteseconazol es el primero de una nueva clase de tetrazoles antifúngicos. Su mecanismo de acción es casi el mismo que el de los triazoles (inhiben la síntesis de ergosterol) pero son más selectivos para la enzima fúngica. |
| Formulación y presentación Cápsulas orales de 150 mg. |
| Actividad ACTIVO – Mayoría de las especies de Candida, incluyendo C. albicans y C. krusei resistentes a azoles, y C. glabrata y C. auris resistentes a equinocandinas. Rhizopus spp. Trichopyton spp. Microsporum spp. Coccidioides spp. Cryptococcus spp. Histoplasma spp. Blastomyces spp. La susceptibilidad reducida o la resistencia pueden estar mediadas por sobreexpresión de bombas de eflujo como CDR1 y MDR1, y por sobreexpresión del blanco de acción del azol, lanosterol 14-alfa-demetilasa (CYP51). INACTIVO – Aspergillus spp. |
| Regimen típico Prevención de la recurrencia de la CVVR: la dosificación requiere de dos dosis de carga de 600 mg el día 1 y 450 mg el día 2; luego 150 mg una vez por semana, hasta un máximo de 11 semanas. También se usa un regimen alternativo que combina oteseconazol y fluconazol. |
| Farmacocinética El oteseconazol se absorbe lentamente durante 5-10 hs; la absorción aumenta cuando se toma con alimentos grasos. Tiene un volumen de distribución muy elevado, un porcentaje de unión a proteínas mayor a 99, una larga vida media de 138 días y no se metaboliza de manera significativa. Las concentraciones pico se alcanzan luego de 6-9 hs y la vida media es de 16-24 hs. Se distribuye ampliamente en la mayoría de los tejidos en concentraciones mucho mayores que las del plasma (4 a 50 veces más), pero no en el cerebro. |
| Interacciones medicamentosas Otesenconazole tiene pocas interacciones documentadas. Es inhibidor del sustrato del transportador del tansportador de la BCRP (proteína de resistencia del cáncer de mama, por su sigla en inglés). Se prevén interacciones con antraciclinas, mitoxantrona, metotrexato, topotecán, irinotecán, estatinas (rosuvastatina), imatinib y otros inhibidores de la tirosin-quinasa, gliburida, nitrofurantoína, dipiridamol, cimetidina, clorotiazida, sulfasalazina, ketamina, prazosina y leflunomida. Pueden ser o no clínicamente significativas. El uso concomitante con rosuvastatina aumentó las concentraciones de esta droga en un 120%. Es posible la exposición aumentada (frecuentemente a través de la absorción aumentada en el intestino) de estas drogas y pueden requerirse ajustes de las dosis. Consultar la base de datos de interacciones. |
| Efectos adversos y toxicidad En ratas embarazadas, oseteconazole resultó en varias anormalidades oculares diferentes a dosis aproximadamente 3.5 veces mayores a la exposición clínica en estado estacionario observado en pacientes tratadas por CVVR. Por este motivo, y considerando especialmente la larga vida media de la droga en el organismo, el otesenconazole está contraindicado en mujeres en edad reproductiva y embarazadas. Los efectos adversos más comunes durante el tratamiento fueron cefaleas y náuseas. Tuvo efectos adversos ligeramente menores que los del fluconazol. |